Modeling biological and chemical sequences is extremely difficult mainly due to the need to handle long-range dependencies and efficient processing of large sequential data. Classical methods, particularly Transformer-based architectures, are limited by quadratic scaling in sequence length and are computationally expensive for processing long genomic sequences and protein modeling. Moreover, most existing models have in-context learning constraints, limiting their ability to generalize to new tasks without retraining. Overcoming these challenges is central to accelerating applications in genomics, protein engineering, and drug discovery, where sequence modeling with precision can lead to precision medicine and molecular biology breakthroughs.
Existing methods are mainly based on Transformer-based architectures, which are strong in representation learning but computationally expensive due to the self-attention mechanism. State-space models like S4 and Mamba have been proposed as alternatives, with improved efficiency in handling long-range dependencies. These models are, however, still computationally expensive and lack flexibility across a broad range of biological modalities. Transformers are robust but afflicted with short context windows, limiting their efficiency in applications involving long-sequence modeling, e.g., DNA analysis and protein folding. Such inefficiencies become a bottleneck to real-time applications and hinder the scalability of AI-powered biological modeling.
To overcome these limitations, researchers from Johannes Kepler University, and NXAI GmbH Austria present Bio-xLSTM, an xLSTM variant specially tailored for biological and chemical sequences. In contrast to Transformers, Bio-xLSTM has linear runtime complexity in terms of sequence length and is much more efficient for sequence processing. Innovations are DNA-xLSTM for genomic sequences, Prot-xLSTM for protein prediction, and Chem-xLSTM for small molecule synthesis. Each variant leverages specialized mechanisms, reverse-complement equivariant blocks in the case of DNA sequences, to better understand sequences. With improved memory components and exponential gates, this innovation enables constant-memory decoding at inference time, hence being highly scalable and computationally efficient.
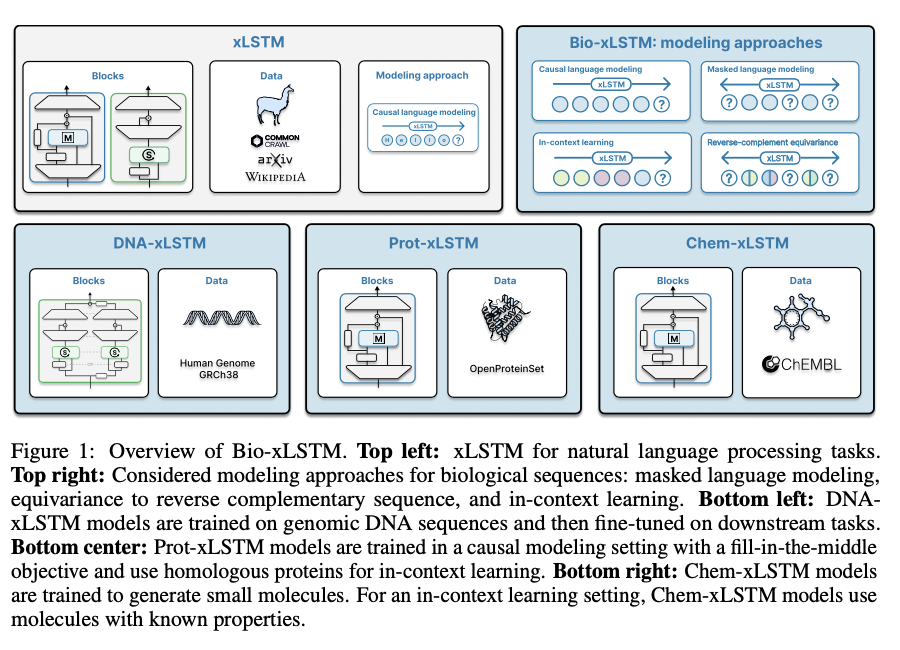
Bio-xLSTM leverages an arsenal of architecture variants that are optimized for various sequence types. DNA-xLSTM uses reverse-complement equivariant mechanisms to take advantage of DNA strand symmetry, and Prot-xLSTM uses homologous protein information for improved representation learning. Chem-xLSTM is aimed at SMILES-based molecule representations and supports in-context learning for synthesizing small molecules. The datasets used are large-scale genomic, protein, and chemical sequence databases, which support effective pre-training and fine-tuning. Training practices are causal language modeling and masked language modeling with context lengths from 1,024 to 32,768 tokens. Each variant is optimized for the respective domain but maintains the xLSTM efficiency advantage.
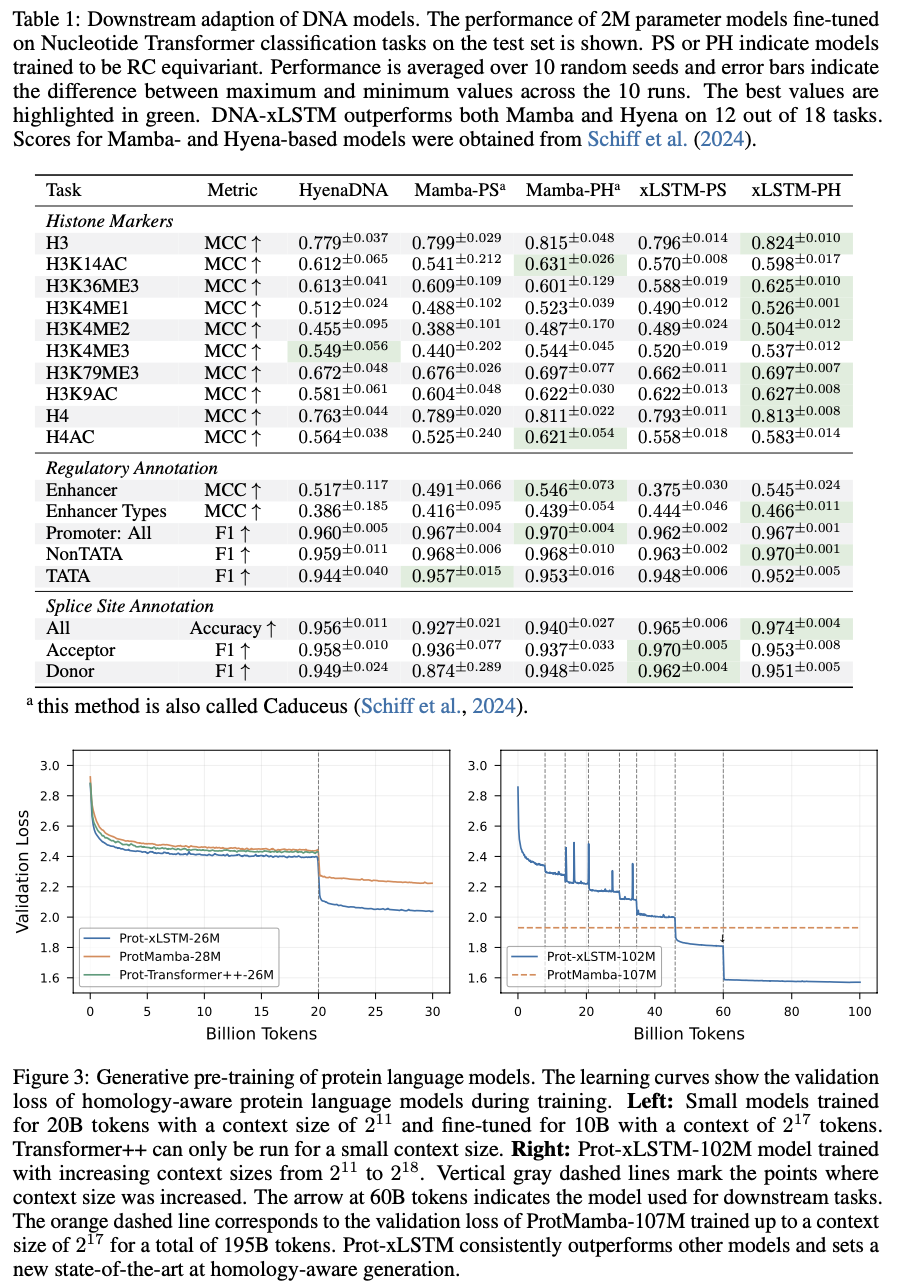
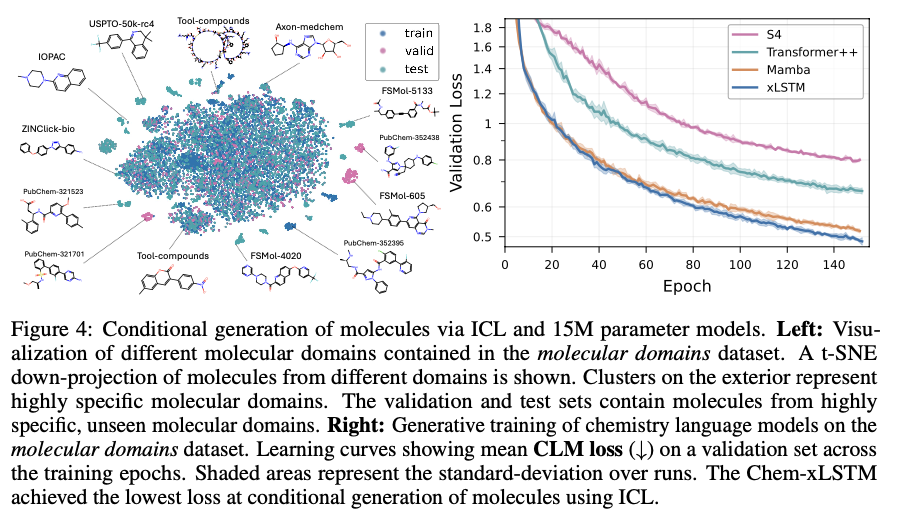
Bio-xLSTM outperforms current models on all genomic, protein, and chemical sequence modeling tasks overall. In DNA sequence tasks, it has lower validation loss than Transformer-based and state-space models and shows higher efficiency in masked and causal language modeling. In protein modeling, it performs better in homology-aware sequence generation with lower perplexity and better long-range dependency adaptation. In chemical sequences, it generates chemically valid structures with high accuracy and outperforms other generative models. These gains in efficiency, accuracy, and adaptability indicate its potential to model various biological and chemical sequences efficiently.
Bio-xLSTM is a game-changer in sequence modeling for biological and chemical applications. By outperforming the computational constraints of Transformers and incorporating domain-specific adaptations, it provides a scalable and highly effective solution for DNA, protein, and small molecule modeling. Its robust performance in generative modeling and in-context learning establishes its potential as a foundational tool in molecular biology and drug discovery, opening the door to more efficient AI-driven research in life sciences.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. Don’t Forget to join our 75k+ ML SubReddit.
🚨 Meet IntellAgent: An Open-Source Multi-Agent Framework to Evaluate Complex Conversational AI System (Promoted)
Aswin AK is a consulting intern at MarkTechPost. He is pursuing his Dual Degree at the Indian Institute of Technology, Kharagpur. He is passionate about data science and machine learning, bringing a strong academic background and hands-on experience in solving real-life cross-domain challenges.
