Generating all-atom protein structures is a significant challenge in de novo protein design. Current generative models have improved significantly for backbone generation but remain difficult to solve for atomic precision because discrete amino acid identities are embedded within continuous placements of the atoms in 3D space. This issue is especially significant in designing functional proteins, including enzymes and molecular binders, as even minor inaccuracies at the atomic scale may impede practical application. Adopting a novel strategy that can effectively tackle these two facets while preserving both precision and computational efficiency is essential to surmount this challenge.
Current models such as RFDiffusion and Chroma concentrate mainly on backbone configurations and offer restricted atomic resolution. Extensions such as RFDiffusion-AA and LigandMPNN attempt to capture atomic-level complexities but are not able to represent all-atom configurations exhaustively. Superposition-based methods like Protpardelle and Pallatom attempt to approach atomic structures but suffer from high computational costs and challenges in handling discrete-continuous interactions. Moreover, these approaches struggle with achieving the trade-off between sequence-structure consistency and diversity, making them less useful for realistic applications in exact protein design.
Researchers from UC Berkeley and UCSF introduce ProteinZen, a two-stage generative framework that combines flow matching for backbone frames with latent space modeling to achieve precise all-atom protein generation. In the initial phase, ProteinZen constructs protein backbone frames within the SE(3) space while concurrently generating latent representations for each residue through flow-matching methodologies. This underlying abstraction, therefore avoids direct entanglement between atomic positioning and amino acid identities, making the generation process more streamlined. In this subsequent phase, a VAE that is hybrid with MLM interprets the latent representations into atomic-level structures, predicting sidechain torsion angles, as well as sequence identities. The incorporation of passthrough losses improves the alignment of the generated structures with the actual atomic properties, ensuring increased accuracy and consistency. This new framework addresses the limitations of existing approaches by achieving atomic-level accuracy without sacrificing diversity and computational efficiency.
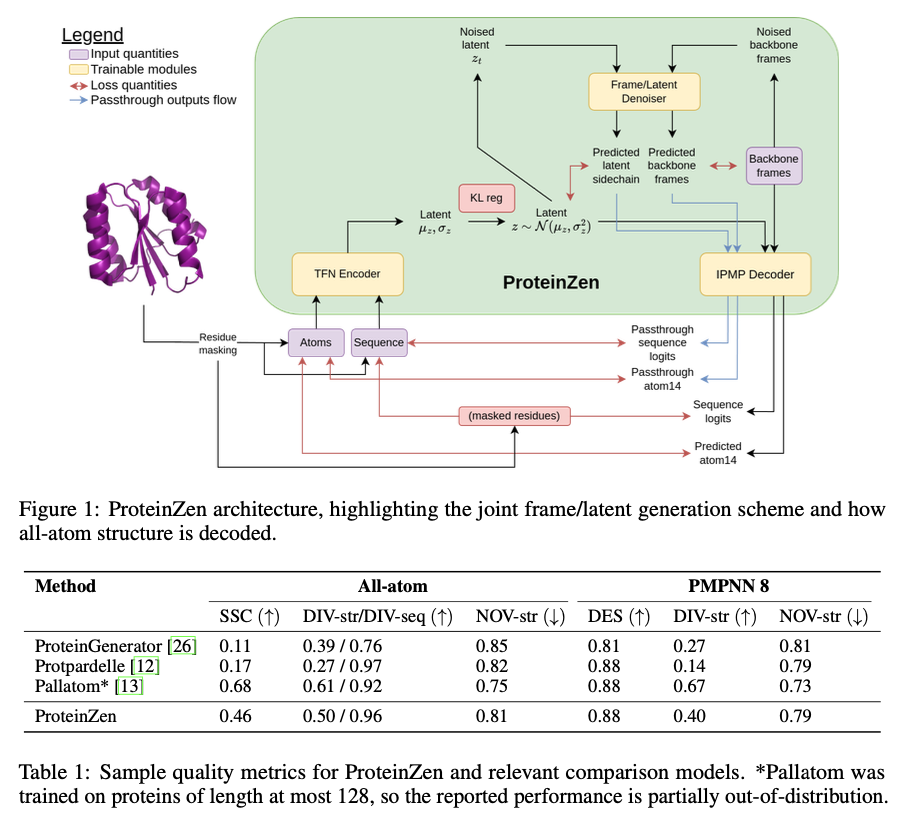
ProteinZen employs SE(3) flow matching for backbone frame generation and Euclidean flow matching for latent features, minimizing losses for rotation, translation, and latent representation prediction. A hybrid VAE-MLM autoencoder encodes atomic details into latent variables and decodes them into a sequence and atomic configurations. The model’s architecture incorporates Tensor-Field Networks (TFN) for encoding and modified IPMP layers for decoding, ensuring SE(3) equivariance and computational efficiency. Training is done on the AFDB512 dataset, which is very carefully built by combining PDB-Clustered monomers along with representatives from the AlphaFold Database that contains proteins with up to 512 residues. The training of this model makes use of a mix of real and synthetic data to improve generalization.
ProteinZen achieves a sequence-structure consistency (SSC) of 46%, outperforming existing models while maintaining high structural and sequence diversity. It balances accuracy with novelty well, producing protein structures that are diverse yet unique with competitive precision. Performance analysis indicates that ProteinZen works well on smaller protein sequences while showing promise to be further developed for long-range modeling. The synthesized samples range from a variety of secondary structures, with a weak propensity toward alpha-helices. The structural evaluation confirms that most of the proteins generated are aligned with the known fold spaces while showing generalization towards novel folds. The results show that ProteinZen can produce highly accurate and diverse all-atom protein structures, thus marking a significant advance compared to existing generative approaches.
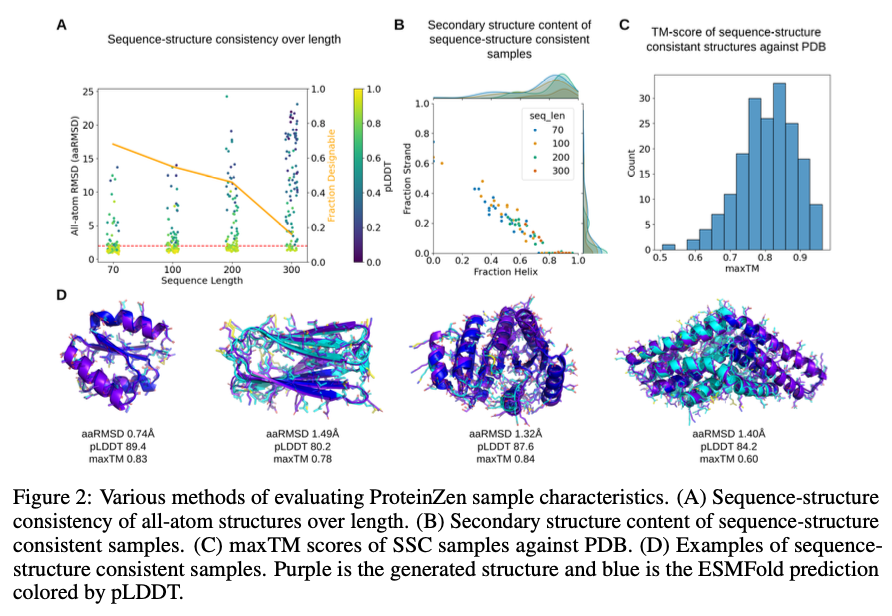
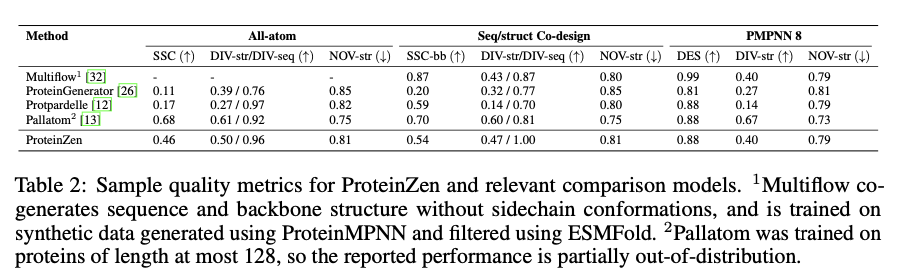
In conclusion, ProteinZen introduces an innovative methodology for the generation of all-atom proteins by integrating SE(3) flow matching for backbone synthesis alongside latent flow matching for the reconstruction of atomic structures. Through the separation of distinct amino acid identities and the continuous positioning of atoms, the technique attains precision at the atomic level, all the while preserving diversity and computational efficiency. With a sequence-structure consistency of 46% and evidenced structural uniqueness, ProteinZen establishes a novel standard for generative protein modeling. Future work will include the improvement of long-range structural modeling, refinement of the interaction between the latent space and decoder, and the exploration of conditional protein design tasks. This development signifies a significant progression toward the precise, effective, and practical design of all-atom proteins.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. Don’t Forget to join our 60k+ ML SubReddit.
Aswin AK is a consulting intern at MarkTechPost. He is pursuing his Dual Degree at the Indian Institute of Technology, Kharagpur. He is passionate about data science and machine learning, bringing a strong academic background and hands-on experience in solving real-life cross-domain challenges.
